
Application Note
Automating the generation and analysis of 3D cell cultures in Matrigel®
Michael Kowalski, Staff Applications Scientist | Beckman Coulter Life Sciences, Indianapolis, IN
Kayla Hill, Cellular Imaging Field Applications Scientist | Molecular Devices, San Jose, CA
Kristin Prasauckas, Manager, Field Applications Scientist | Molecular Devices, San Jose, CA
Tara Jones-Roe, Marketing Manager | Beckman Coulter Life Sciences, Indianapolis, IN
Introduction
Researchers are turning to three-dimensional (3D) cell culture as a way to increase the biological relevance of their disease models and predictive value of drug studies1. These 3D models typically increase the level of cell-cell interaction and reduce or eliminate the dominant cell-plastic interaction seen in standard monolayer cultures. There are a number of ways of generating 3D cell cultures, each with unique advantages and challenges, many of which automation can help overcome.
One common way of culturing cells in three dimensional space is through the use of extracellular matrix-based hydrogels such as Matrigel. Differences between Matrigel and 2D cell cultures can be readily seen by their different cell morphologies (Figure 1), cell polarity, and/or gene expression (reviewed in 2Baker and Chen). Matrigel can also enable studies on cell migration and 3D structure formation, such as endothelial cell tube formation in angiogenesis studies3,4.
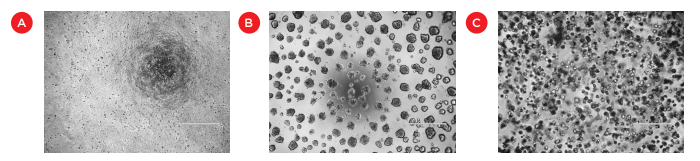
Figure 1. Images of HCT 116 cells grown in standard monolayer cultures (A), on the surface of Matrigel (B), or embedded within a layer of Matrigel (C). Bar = 500 μm.
One of the unique properties of Matrigel that makes it difficult to work with is that it is a liquid only at cold temperature and solidifies into a gel at room temperature or 37°C. Cells are typically grown either on top of a coating of Matrigel or embedded within a thicker layer of Matrigel (Figure 1), with supplemental media typically added on top of the hydrogel layer following solidification. Assaying cells grown in Matrigel can also be problematic. Options include using plate readers or imaging devices to interrogate the cells while still in the matrix or slowly dissolving the Matrigel to enable the processing of cells in suspension or lysates. All of these steps have significant challenges when performed manually and we will describe how we have utilized automation to plate, treat, and prepare Matrigel cultures for analysis. In addition, automation enabled the use of 384-well plates, thereby introducing significant cost savings by reducing the amount of Matrigel and other expensive reagents used per condition.
Summary
- Automated the plating, drug treatment, and staining of Matrigel cultures on the Biomek FX
- P
- Workstation
- Temperature control positions maintained Matrigel in liquid form and induced gelatinization
- Optimized pipetting achieved cost savings through reduced Matrigel usage
- Measured cell growth and apoptosis induction in Matrigel cultures using:
- SpectraMax i3X Multi-Mode Detection Platform with SpectraMax MiniMax 300 Imaging Cytometer
- ImageXpress ® Micro Confocal High-Content Imaging System
Automated Matrigel Plating and Culture
The Matrigel workflow was automated on a Biomek FXP Workstation (Beckman Coulter) with a 96-channel head and Span-8 pipettors (Figure 2). The flexible Biomek Workstation was configured with numerous temperature-controlled Peltier positions to maintain the Matrigel in liquid form and to heat the assay plate to induce gel polymerization. The 96-channel head featured enhanced multichannel selective tip pipetting (EST), which allows any pattern of tips to be used. Sterility of the automated cell manipulations was protected through the use of sterile Biomek pipette tips and a HEPA-filtered enclosure and no contamination was detected even in the absence of antibiotics in the cell media.
Cell Plating
Phenol red-free Matrigel (Corning) thawed on ice was diluted to 6 mg/mL with FluoroBrite DMEM medium (Thermo Fisher Scientific) and for embedded cultures, HCT 116 cells were added to a concentration of 100,000 cells/mL. The Matrigel (+/- cells) was added to a single column of a deep well plate that was maintained at 4°C on a shaking Peltier device with a deep well adapter. 25 μL was added in column-wise fashion to wells of a 384-well black-walled clear bottom plate (Greiner) using the multichannel EST function. This assay plate was then heated to 37°C on a static Peltier for 30 minutes to solidify the Matrigel. 75 μL FluoroBrite DMEM medium containing 10% FBS was then added to each well. For cultures grown on a surface of Matrigel, HCT 116 cells were added to the supplemental media rather than to the Matrigel. Final cell plating numbers were 2500 cells/well for embedded cultures and 8000 cells/well for surface cultures.
As described above, there are numerous challenges when plating Matrigel cultures. Automation was able to consistently position the pipette tip 0.3 mm above the well bottom such that the low volume of Matrigel would spread to cover the entire well. Manual pipetting required making contact with either the well bottom (potentially scratching and/or affecting the transfer volume) or the side of the well, resulting in the Matrigel droplet adhering to the side and failing to cover the well. The only way we could achieve consistent well coverage through manual transfer was by doubling the Matrigel volume to 50 μL, thereby doubling the cost of the experiments.
Another advantage to automated plating was the ability to use slow aspirate, dispense, and mix speeds to avoid generating bubbles in the Matrigel solution. We also aspirated a small additional volume of Matrigel into the pipette tip to avoid introducing a bubble when dispensing to the assay plate. Although they are a frequent complaint with Matrigel cultures, no bubbles were seen in cultures immediately following automated plating. Cultures were stored at 37°C in a 5% CO2 incubator for various times before treatment and/or analysis.
Cell Growth Assays
To measure initial cell growth for Matrigel surface cultures, 100 μL XTT reagent was added to wells each day over the first three days of culture. Absorbance was measured at 475 nm and 660 nm using the SpectraMax i3X Multi-Mode Detection Platform ("SpectraMax i3X reader", Molecular Devices, Figure 2). A linear growth rate was seen (R2 = 0.985) indicating consistent growth of 8000 cells over this time frame (Figure 3A). The slight plateauing of the absorbance at day 3 could reflect a slowing of growth but is more likely an indication that we are approaching the absorbance maximum for the XTT assay and should shorten the reagent incubation time for additional time points.

Figure 2. A Biomek FXP Workstation (left) was used for plating, treating, and staining Matrigel cultures. These cultures were then analyzed with either the SpectraMax i3X reader with MiniMax cytometer (center) or the ImageXpress Micro Confocal system (right).
As the XTT assay is an endpoint assay, we also sought to track cell growth in a label-free manner. We utilized the SpectraMax i3X reader with a SpectraMax MiniMax 300 Imaging Cytometer ("MiniMax cytometer", Molecular Devices) to measure the percentage of the well occupied by surface colonies (“field” analysis setting). Changes in confluence of surface colonies could readily be detected over time (data not shown) despite the slight concavity of the plated Matrigel preventing a clear plane of focus. More importantly, the colony morphology of these cultures are such that a doubling in cell numbers may not give a two-fold increase in confluence. The XTT assay may provide a more accurate representation of the increase in cell number, but the imaging approach can act as a reasonable surrogate to ensure that cell growth is occurring in a steady fashion while still allowing the cells to be used for an experiment at a future date.
The lack of a focal plane is even more challenging when imaging cultures embedded in Matrigel. However, we were able to use the field measurement on the MiniMax cytometer to again see differences between wells that received different cell plating numbers as well as track growth over time. Figure 3B shows well images acquired and analyzed on the MiniMax cytometer to identify regions that have cells present. One can see the increasing amount of masked cells over a three day period. This increase and the level of confluence of other cell plating numbers is plotted in Figure 3C. Again, while perhaps not a direct measurement of the increase in cell numbers, the high level of linearity in the growth curves (R2 values between 0.968 and 0.999) and the clear separation between plated cell numbers suggest this approach can be used to track cell growth in embedded Matrigel cultures in a label-free manner.
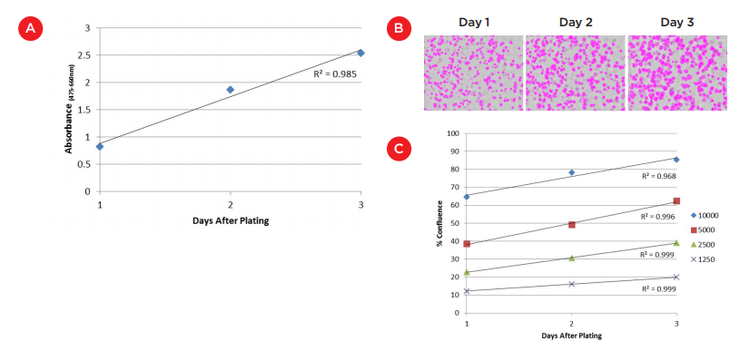
Figure 3. Analysis of cell growth on/in Matrigel. A) XTT assay of 8000 cells grown on the surface of Matrigel over three days. Absorbance measurements were acquired on the SpectraMax i3X reader after 4 hours of incubation with XTT reagents. B) Confluence measurements of HCT 116 cells plated within Matrigel as measured using the field analysis on the SpectraMax i3X reader with MiniMax cytometer. The images show the increase in the masked area of 2500 cells over three days. C) Graph of confluence measurements for various starting cell numbers. Change in confluence over three initial days of culture was highly linear for all cell numbers (R2 0.968 to 0.999).
Compound Treatment and Apoptosis Analysis
Compound Dilution and Addition
One of the goals of 3D culture is to determine how cells respond to stimuli in more biologically relevant systems. We sought to determine how colon cancer cells grown in Matrigel respond to apoptosis inducers. Five days after plating HCT 116 cells embedded in Matrigel, apoptosis-inducing compounds were diluted in NucBlue® Live ReadyProbes® Reagent (Thermo Fisher Scientific) to the following concentrations: 10 μM staurosporine, 20 μM camptothecin, and 375 μM 5-fluorouracil. Compounds were diluted in NucBlue due to the need for long incubation times to stain cell nuclei in Matrigel. These solutions were added to the first row of a 96-well round-bottom plate and the Span-8 pipettors transferred NucBlue containing 1% DMSO to the remaining rows of the plate. The compounds were then serially diluted 1:3 in a row-wise fashion using the EST, leaving the final row as a negative control (DMSO alone).
10 μL of media was aspirated from Matrigel wells starting from the 100 μL liquid height and following the level of the liquid during the aspiration. Not only did this ensure that we did not disrupt the Matrigel, but it enabled wells that may have lost media due to evaporation (i.e. edge wells) to return to the same volume as the rest of the plate, since air would be aspirated until the pipette tip reached liquid. This volume normalization ensured accurate final compound concentrations across the plate following the addition of 10 μL compound dilutions to replicate wells.
Apoptosis Assays
24 hours after compound treatment, 10 μL CellEvent® Caspase-3/7 Green (Thermo Fisher Scientific) was added and cultures were incubated at 37°C for 6 hours to stain apoptotic cells. Cell images were acquired with an ImageXpress® Micro Confocal High-Content Imaging System ("ImageXpress Micro Confocal system", Molecular Devices, Figure 2). Wells were imaged at 4X magnification using DAPI and FITC filters. This lower magnification allowed the entire well to be imaged and by acquiring Z-stacks, we were able to visualize embedded cells in three-dimensional space, as shown in Figure 4A. Figure 4B shows the two-dimensional projection of z-stacks taken for replicate wells of the top 5 dilutions for each inducer. These projections allowed for the enumeration of total cells (blue nuclei) and caspase-positive cells (green) embedded throughout the thick 2 mm Matrigel layer to determine the relative level of apoptosis induced by each compound.
The percentage of caspase-positive cells at each concentration (average of two replicate wells) is shown in Figure 4C. Both staurosporine and camptothecin reached maximal toxicity and showed a traditional dose-response curve, thereby allowing the calculation of an IC50 for HCT 116 cells embedded in Matrigel. With an IC50 of 6 nM, staurosporine was roughly 10-fold more toxic to HCT 116 cells than camptothecin which had an IC50 of 80 nM. 5-fluorouracil gave a much weaker effect and while 50% toxicity was reached near the maximal concentration of 3.75 μM, any IC50 value would likely be unreliable. This weaker effect for 5-fluorouracil has also been seen in our previous work with 3D models of HCT 116 cells.
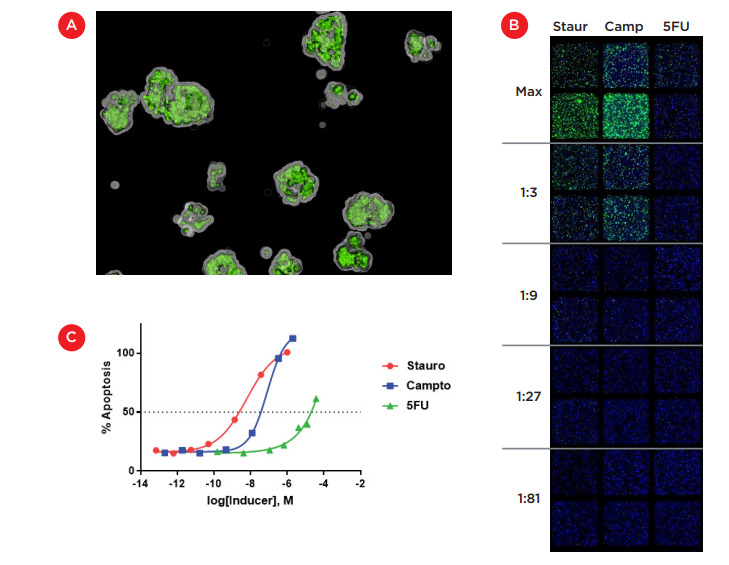
Figure 4. Induction of apoptosis in Matrigel-embedded cultures. A) Image acquired using the ImageXpress Micro Confocal system illustrating the three-dimensional colonies stained with a fluorescent caspase 3/7 substrate as a marker of apoptosis. B) Montage of 2D projections of Z-stacks acquired at 4X magnification with the ImageXpress Micro Confocal system. Replicate wells were treated with dilutions of staurosporine, camptothecin and 5-fluoruracil (top 5 dilutions shown) and stained for cell nuclei (blue) and apoptosis (green). C) Dose-response curves generated from imaging data for each compound.
Discussion
Matrigel cultures can be highly valuable for numerous studies. While we demonstrated a compound screen for apoptosis induction in 3D-cultured colon carcinoma cells, Matrigel also allows for other assays that require three dimensional space such as vascularization or neurite outgrowth studies5. However, the challenges to utilizing Matrigel assays are significant. We have shown how finely controlled pipetting speeds and positions coupled with temperature regulation allowed automation to overcome a number of these obstacles. Just one example is how automation enabled the miniaturization of these assays to a 384-well format and led to significant cost savings by reducing Matrigel usage by 50% compared to manual plating or a 96-well format. This smaller format also requires less staining reagents and allows faster confocal imaging, as multiple fields of view would be necessary to capture the entire well of a 96-well plate.
The flexibility of the Biomek Workstation also enables diverse workflows or throughputs. For example, we performed the heating of the Matrigel assay plate on a single Peltier position, but if numerous plates needed to be processed simultaneously, one has the ability to integrate additional Peltiers or an incubator. Higher throughput applications are also assisted by integrating plate and tip storage and analyzers such as the SpectraMax i3X reader with MiniMax cytometer or ImageXpress Micro Confocal system used here.
Additional observations were made during this work that warrant consideration. First, while no bubbles were introduced during Matrigel plating, some small bubbles formed on the edges of the wells during gelatinization. Interestingly, this seemed to be due to the plate characteristics as this delayed bubble formation was also seen with PBS addition in the absence of Matrigel. Regardless, automated plating consistently resulted in less bubbles than manual cultures, despite the use of narrow pipette tips to access the 384-well plates.
When plating embedded cultures, some cells will naturally settle to the well bottom before gelatinization occurs and these cells may behave like monolayer cultures, such as showing a spread morphology. The ideal option might be first plating a layer of Matrigel alone, followed by the cells embedded in additional Matrigel. However, this roughly doubles the cost of Matrigel required per plate due to the minimum volume required to fully cover the well.
One way of reducing these 2D cells is to accelerate the gelatinization process by preheating the assay plate on the Peltier position. This would also minimize any differences across a plate if one is plating in a column-wise fashion rather than all 384-wells at once. However, this approach seemed to somewhat increase the number of bubbles on the edges of the well. While this would not likely affect the plating volume or cell growth, nor be detected in well-level assays such as the XTT assay bubbles may interfere with image analysis, particularly at higher magnifications. Normalizing stained cells to total cells can help reduce this effect, however, by acquiring Z-stacks with the ImageXpress Confocal system, we can essentially image the entirety of the Matrigel well and dramatically reduce the likelihood that the presence of a bubble would result in inaccurate results. An even greater benefit to the Z-stack feature is the ability to control the height at which the acquisition begins. By starting the acquisition above the well bottom, one can eliminate any signal from the 2D cells and ensure only 3D cells are included in the analysis.
Another challenge to imaging cells in Matrigel was that a number of dyes gave high levels of background and/or required lengthy incubation times. This was not unexpected and is why numerous protocols suggest fixing the cells, blocking the wells, and washing the wells after staining. We found we were able to stain live cells with lengthy incubations with the NucBlue nuclear dye, hence this reagent was added at the time of compound treatment. We also determined that the caspase 3/7 reagent gave less background than a number of other live/dead reagents (data not shown). This may be due to the fact that this reagent must be enzymatically cleaved to fluoresce, therefore, binding to Matrigel should not result in significant background signal.
While Matrigel is just one method for studying 3D cultures, we have shown how this method may be used to measure the susceptibility of cancer cells to apoptosis inducers in a more physiologically relevant system than 2D culture. Automation of the plating, treatment, and staining of these cultures reduced time at the bench and reagent costs while also overcoming the difficulties of this temperature-sensitive reagent to achieve consistent cell growth and measurable dose response curves.
References
- Kim YJ, et. al. (2009). Three-dimensional gastric cancer cell culture using nanofiber scaffold for chemosensitivity test. Int J Biol Macromol. 45(1):65-71.
- Baker BM and Chen CS. (2012). Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci. 125(Pt 13):3015-24.
- Chen J, et. al. (2016). C-reactive protein can upregulate VEGF expression to promote ADSC-induced angiogenesis by activating HIF-1α via CD64/PI3k/Akt and MAPK/ERK signaling pathways. Stem Cell Res Ther. 7(1):114.
- Mou Y, et. al. (2016). OCT4 Remodels the Phenotype and Promotes Angiogenesis of HUVECs by Changing the Gene Expression Profile. Int J Med Sci. 13(5):386-94.
- Semina E, et. al. (2016). Urokinase and urokinase receptor participate in regulation of neuronal migration, axon growth and branching. Eur J Cell Biol. 95(9):295-310.
Learn more about the ImageXpress Micro Confocal High-Content Imaging System >>